Advancing Quantum Frontiers in Molecular and Materials Science





We are currently working on four exciting areas of research at the cutting edge of quantum science and physical chemistry:
Quantum Control of Electrons in Matter
 A long term goal in our group is to develop useful schemes to control matter at the level of electrons with ultrafast laser sources. The reason why we focus on electrons is because they determine the optical, charge transport, and, ultimately, the chemical reactivity of matter. The reason to focus on lasers as a source of control is because they allow for the manipulation of matter on ultrafast –femto to attosecond– time scales, something that is not achievable by conventional means such as applied voltages, thermodynamic or chemical control.
A long term goal in our group is to develop useful schemes to control matter at the level of electrons with ultrafast laser sources. The reason why we focus on electrons is because they determine the optical, charge transport, and, ultimately, the chemical reactivity of matter. The reason to focus on lasers as a source of control is because they allow for the manipulation of matter on ultrafast –femto to attosecond– time scales, something that is not achievable by conventional means such as applied voltages, thermodynamic or chemical control.
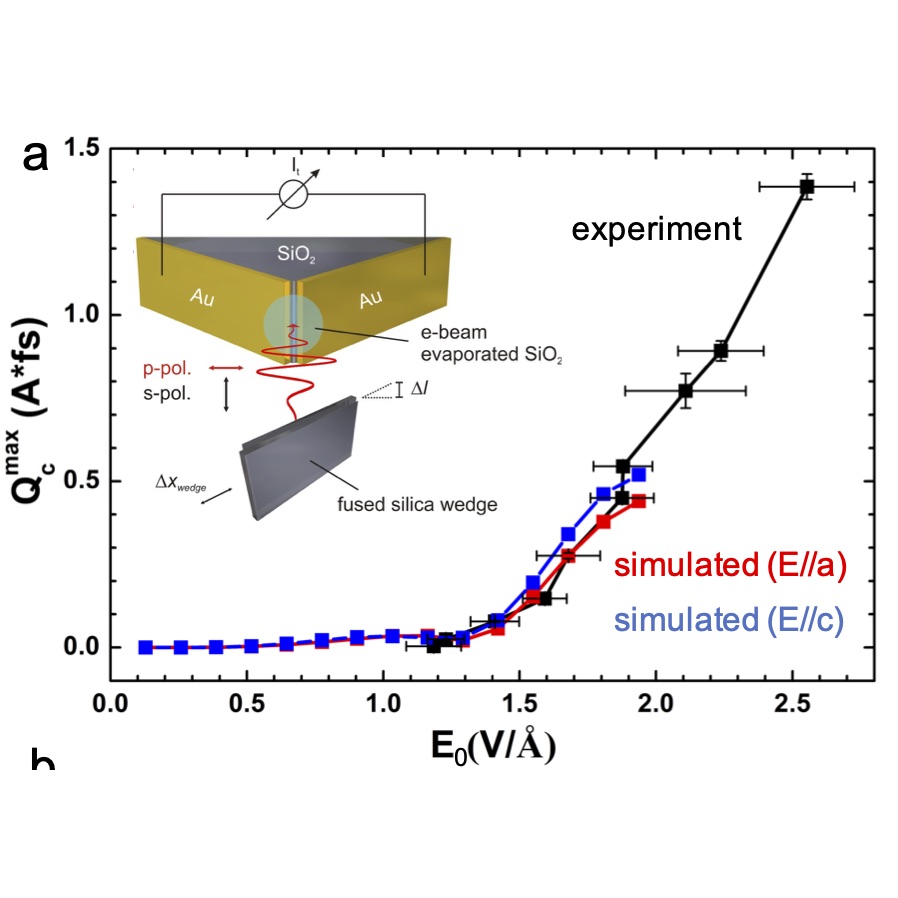
The latest advances in laser technology now enable the generation and control of few-cycle lasers in the IR and UV/Vis. Using such pulses, it is now possible to apply laser fields with amplitudes of ~1-10 V/nm before the emergence of dielectric breakdown. At those intensities, the incident light can dramatically distort the electronic structure of matter as the strength of the light-matter interaction becomes comparable to the strength of chemical bonds, thus opening exciting opportunities to manipulate electron dynamics and to create laser-dressed materials with emergent physico-chemical properties that can be triggered on-demand.
We are pioneering three general strategies for the dynamical quantum control of materials:
 Ultrafast Laser-Driven Currents (1, 2, 3, 4, 5)
Ultrafast Laser-Driven Currents (1, 2, 3, 4, 5)
We develop strategies to generate bursts of electricity on femtosecond time scales along nanoscale junctions using ultrafast laser pulses. The initiative offers the promise of creating a new time-standard for the generation of currents that can be the basis for electronics and information processing at petahertz frequencies which is 5-6 orders of magnitude faster than conventional electronics.
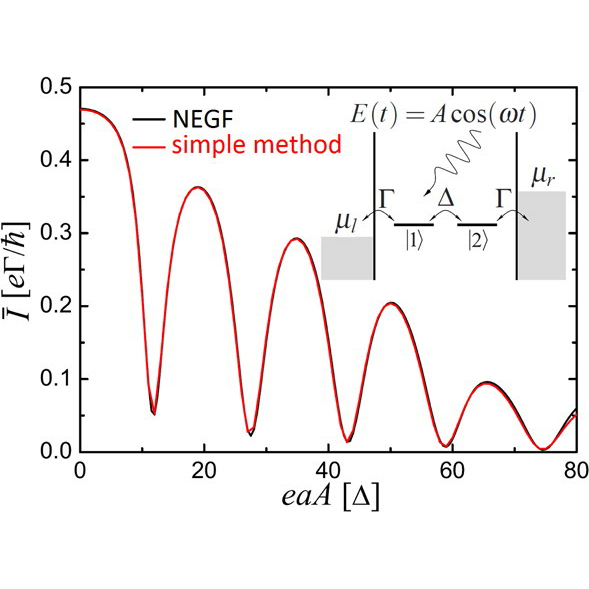
As far as we know, we were the first to propose a strategy to drive currents using the subcycle structure of light (1, 2). The use of the subcycle structure of light makes it possible to control electrons on attosecond timescales even with lasers with femtosecond envelopes. Most recently, in collaboration with the group of Peter Hommelhoff we were able to disentangle the laser-induced current into contributions due to real and virtual charge carries and use this augmented control landscape to demonstrate logic gates that operate at the petahertz limit! (3)
Very interestingly, this type of experiments can also be used to measure electronic coherences through a purely electronic observable! (4)
This direction also motivated us to develop a simple and accurate method to model time-dependent transport in which the leads are incorporated explicitly as mesoscopic contacts that are made to behave like macroscopic contacts (5).
Stark Control of Electrons across Interfaces (SCELI) (6, 7, 8, 9)
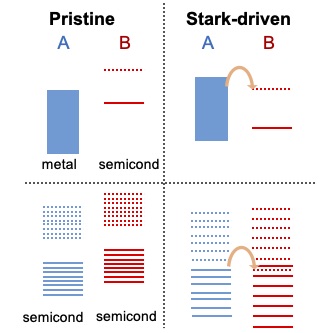
We pioneer schemes to control electron dynamics at interfaces using the Stark effect. While the Stark effect had been shown to be useful to control nuclear dynamics in molecules, our contribution has been to demonstrate its generality and utility to control electron dynamics. The reason why the Stark effect is of interest in the dynamical control of electrons is because it is robust to decoherence. Our control scheme, SCELI, uses non-resonant lasers of intermediate intensity (non-ionizing but whose effects cannot be captured by finite-order perturbation theory) to distort the electronic structure of matter through Stark shifts and create transient resonances between the electronic levels of two adjacent materials A and B. When the level of material A is occupied and the one in B empty, these transient crossings open quantum tunneling pathways for interfacial A to B charge transfer. The SCELI rate of charge transfer is faster than those offered by resonant photoexcitation routes as it is controlled by the subcycle structure of light, and can be used when resonant routes for charge transport do not exist.
Laser-Dressed Materials (10, 11)
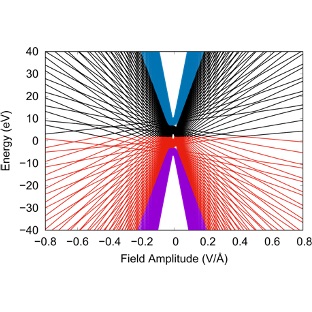
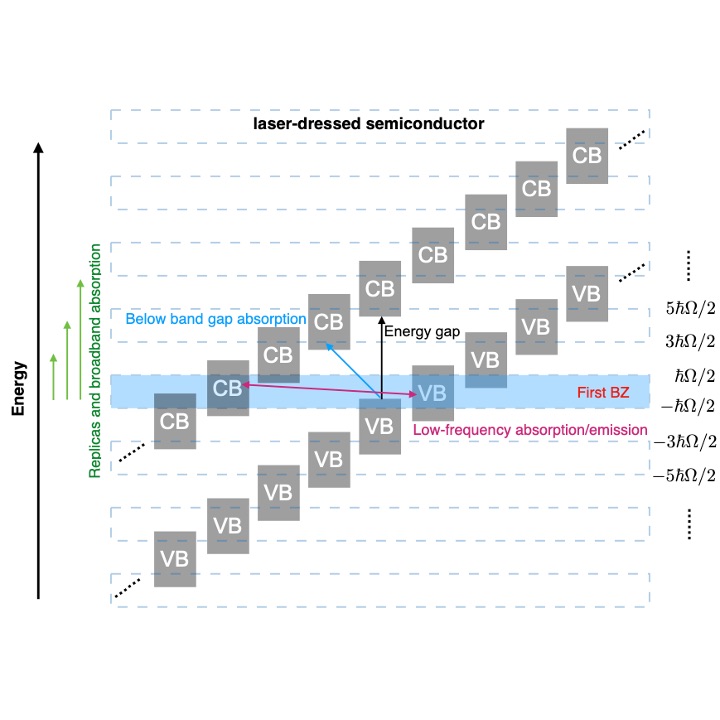
Strong light-matter interactions can also be employed to endow matter with unique physico-chemical properties. These laser-dressed non-equilibrium functional materials are based on deeply optoelectronic effects in which optical fields control their electronic properties. To catalyze the development of laser-dressed materials with “on-demand” properties that are tunable on an ultrafast timescale, it is essential to understand how these materials absorb light, transport charge and in general respond to external stimuli. The theoretical challenge is to generalize the usual response theory that is only applicable for matter near thermal equilibrium to this highly nonequilibrium situation.
We have clarified the linear optical response of molecules and solids dressed with light of arbitrary frequency and intensity. This was done by combining Floquet theory to exactly capture the effect of the dressing laser pulse, and first order perturbation theory to capture the effect of a weak laser that probes the ability of these materials to absorb light. Our main technical advance has been to isolate computationally tractable expressions for the optical response of laser-dressed molecules and solids. Our main discovery has been to demonstrate that the natural states to understand laser-dressed matter are the Floquet modes. Using this theory, we have shown that non-resonant light can transiently turn transparent materials into broadband absorbers, and have isolated distinct purely optical signatures of the Floquet states. Our absolute favorite is the opening of low frequency transitions that emerge due to hybridization (!) of the Floquet modes.
Quantum Coherence in Matter: Tackling the Decoherence Challenge
 A long-term goal in our group is to advance our fundamental understanding of the mechanisms by which quantum coherence is lost in matter and our ability to accurately model the dynamics of open quantum systems that leads to such decoherence. We tackle the basic questions in molecular decoherence research, such as: How fast is the decoherence? What are the main mechanisms for coherence loss? How to quantify and model the decoherence? How can the decoherence be mitigated or exploited?
A long-term goal in our group is to advance our fundamental understanding of the mechanisms by which quantum coherence is lost in matter and our ability to accurately model the dynamics of open quantum systems that leads to such decoherence. We tackle the basic questions in molecular decoherence research, such as: How fast is the decoherence? What are the main mechanisms for coherence loss? How to quantify and model the decoherence? How can the decoherence be mitigated or exploited?
Our work is motivated by the fact that preserving quantum coherence is essential to harness the quantum features of matter as needed to fuel the next technological revolution in computing, sensing and communication. Protecting coherences is also needed to unshackle chemical process from the constraints of thermal Boltzmann statistics as needed to enhance molecular function through coherence, develop novel quantum control routes, and design enhanced spectroscopies. Understanding molecular decoherence is also key in our elementary description of photophysics, photochemistry, molecular interferometry, and charge and energy transfer, and in the development of useful approximate theoretical methods to follow the quantum dynamics of molecules.
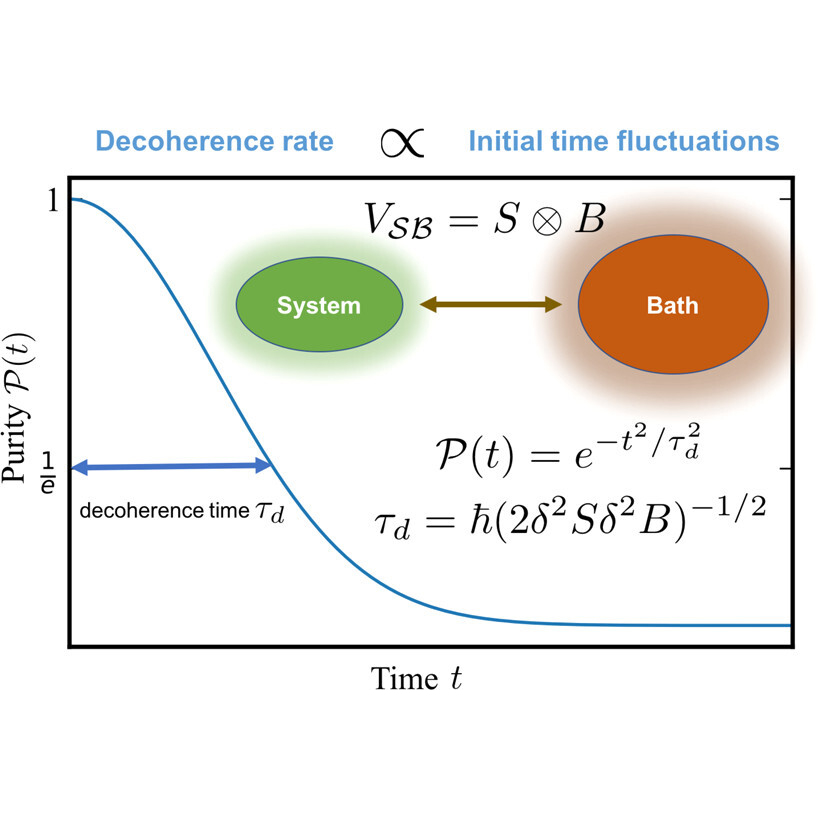
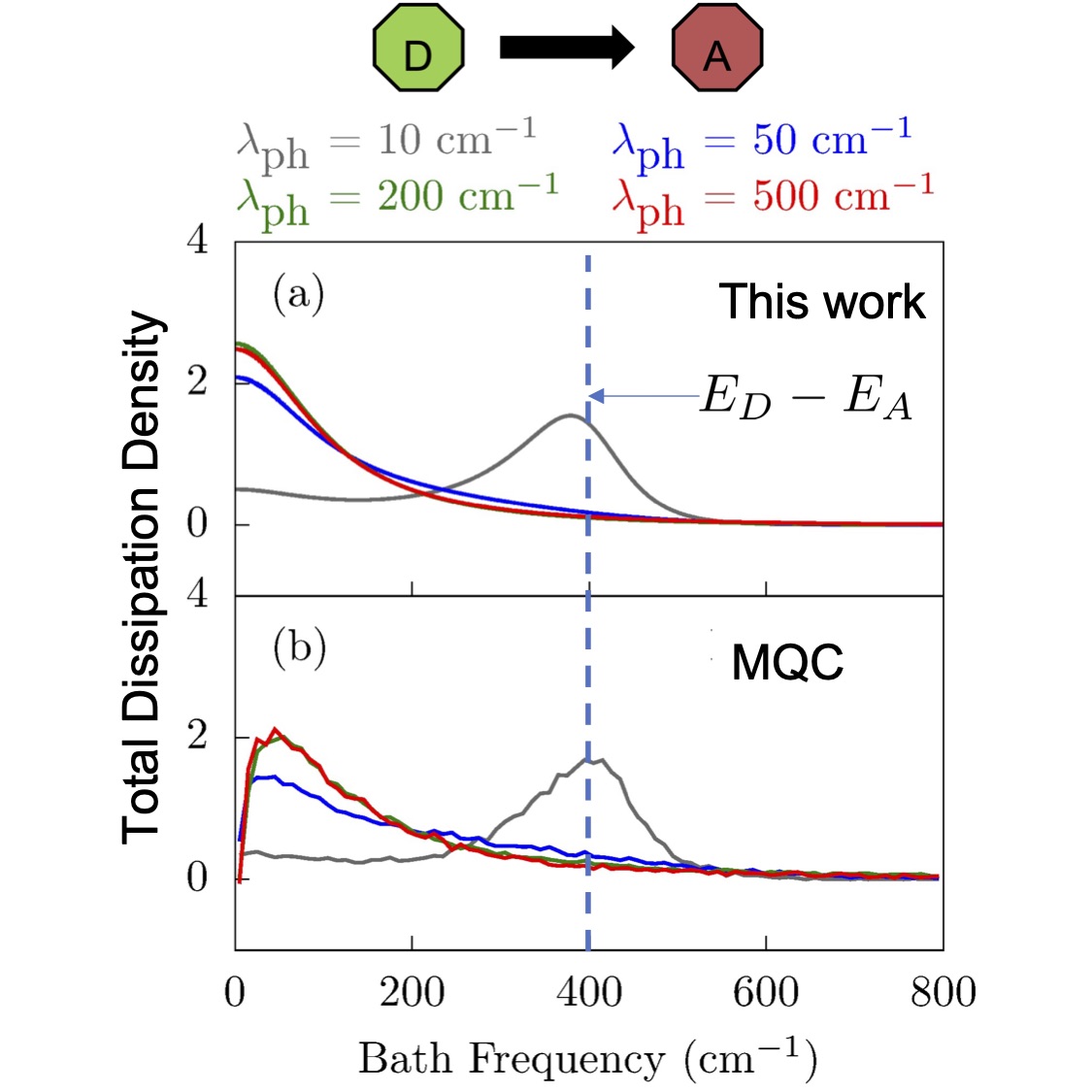
An important result by our group has been the isolation of a universal theory of early-times decoherence time scales (see 1) that makes it possible to identify decoherence mechanisms and estimate the time it takes for coherence to be lost without explicitly propagating the dynamics. We have used this approach to develop a generalized theory of molecular electronic decoherence (see 2) that revealed that nuclear-induced dephasing, electronic transitions and their interference contribute importantly to the decoherence. These insights led us to develop a quantum control scheme to manipulate the decoherence via laser photoexcitation.(see 3) Most recently, they led us to discover that by hybridizing molecular states with those of quantum light in the context of optical cavities it is possible to create optically accessible subspaces with protected coherences with lifetimes that are orders of magnitude longer than those of the bare molecule and tunable by varying cavity frequency! (see 4)
At a computational level, we have modeled the decoherence through quantum dynamics simulations where the bath is taken into account either explicitly or implicitly through quantum master equations. In particular, through explicit simulations we clarified (5) fundamental issues about decoherence (such as the influence of preparation, nuclear mass and anharmonicities, electronic correlations) that require detailed insights into the system-bath entanglement that leads to decoherence that are inaccessible in quantum master equations.
We also investigated many-body aspects of the electronic decoherence (do electronic correlations protect electrons from the decoherence?) and, in doing so, opened a path to use approximate electronic structure theories to model exact decoherence dynamics in molecules. (6)
Another important result by our group has been to clarify the extent to which classical noise models, which are popular but approximate ways to introduce and think about decoherence, can be used to mimic quantum decoherence (7) Most recently, we have extended our decoherence studies into quantum transport, and clarified the importance of incoherent routes to electron transport across molecular junctions immersed in thermal environments.(8) In the process, we clarified the utility and tested the limits of the Landauer formula, which is a central and widely used formula in molecular junctions, for modeling transport through molecules in solution.
Most recently, we have been working on developing a theory of dissipation pathways that now enable us to understand how the structure of the environment leads to coherence loss (see 9).
Quantum Simulation (1)

Computing the quantum dynamics of molecules in condensed phases with high precision is a central challenge in chemistry. Tackling this problem exactly with conventional computers using state of the art methods is a formidable task as it involves solving the Liouville von Neumann (LvN) equation for a molecule interacting with a macroscopic environment that can operate at disparate timescales with varying interaction strengths to the system (thus highly structured), remember the dynamical history of the system (thus non-Markovian), and lead to both energy relaxation, loss of quantum coherence and environment-mediated interactions (thus quantum many-body). In addition to developing new methods in quantum dynamics, we are developing analog quantum simulators of condensed phase chemical dynamics.In this approach, instead of trying to solve the LvN using digital computation, the problem of physical interest is mapped onto a highly controllable experimental setup and Nature is allowed to do the computations.
The quantum hardware that we are investigating for analog quantum simulation are gate-defined semiconductor quantum dots connected to quantum electronic circuits. The theoretical challenge is to establish useful and accurate maps between the Hamiltonian of the target system into the quantum hardware. In our approach, the quantum dots are used to model the system, while arrays of quantum electronic circuits exactly represent the bath. We have been able to show this system can accurately model open quantum dynamics of chemical interest with simulation fidelities > 99.99\% even in the presence of the natural noise of the quantum hardware. The approach can exactly capture the effects of highly structured non-Markovian quantum harmonic environments to all orders in the system-bath interaction, and offer clear potential advantages over conventional and even quantum computation.
Molecular Electronics and Quantum Transport
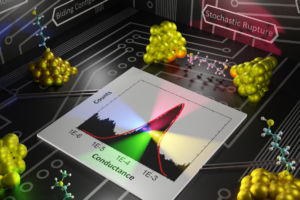
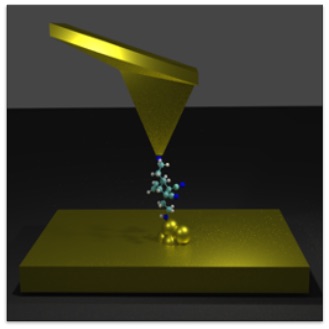
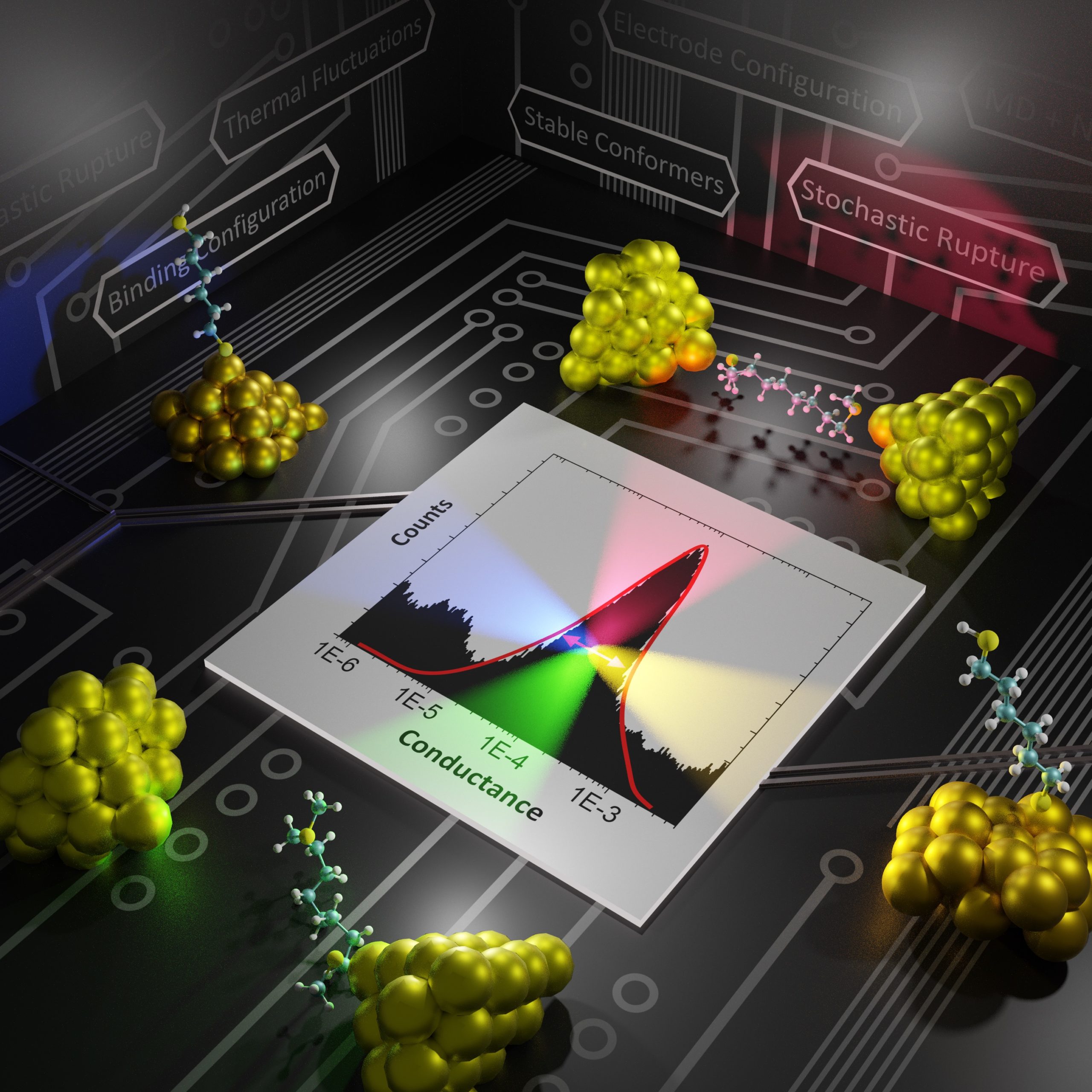
Molecular electronics experiments have emerged as a powerful and versatile platform where voltages, force, and light can simultaneously be applied and used to investigate chemistry and physics at the single-molecule limit. In particular, this class of experiments enable us to interrogate how molecular structure determines charge transport. Our group is developing useful atomistic modeling strategies to capture the two main experimental techniques in this field: ultra-high vacuum STM experiments and room-temperature break junction experiments. We are very interested in modeling and proposing experiments that simultaneously measure force and conductance on single-molecules, as we believe this can become the basis of a useful multidimensional spectroscopy that operates at the single-molecule limit. These are our advances:
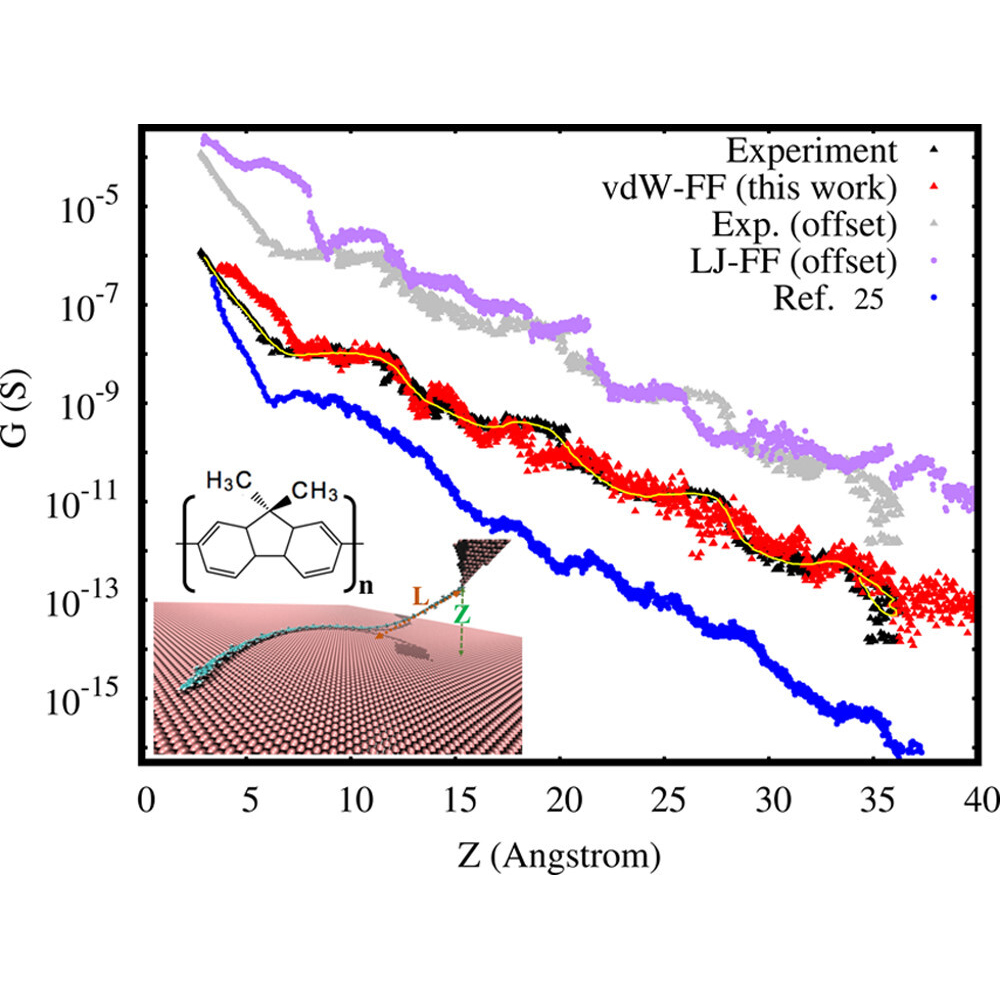
Modeling STM Experiments: We developed an accurate force field for metal-molecule interactions that now enable us to model STM experiments on experimentally relevant system sizes and time scales (see 1 and 2). Once we are able to establish contact with experiments, the simulations offer an atomically sharp microscope of the molecular dynamics!
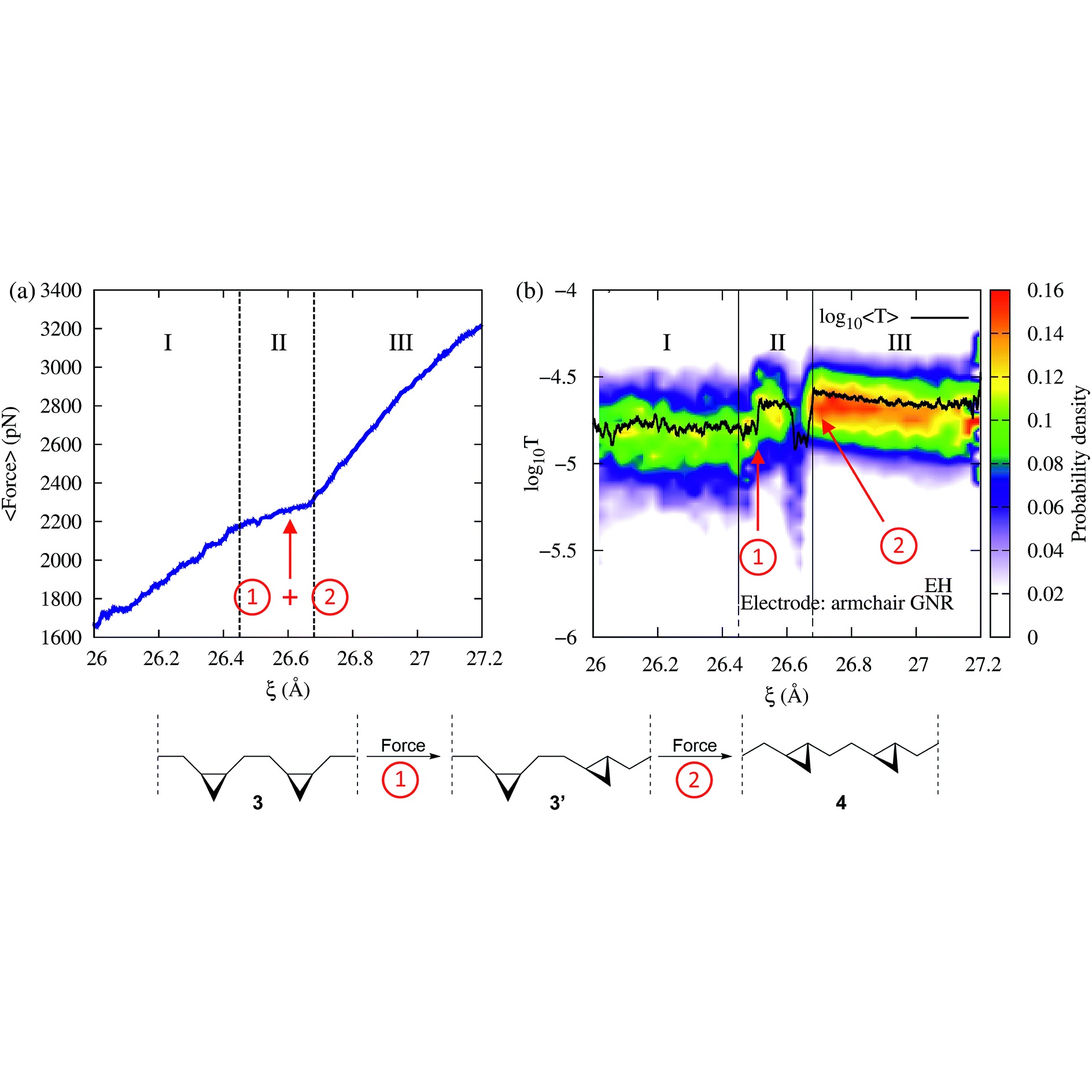
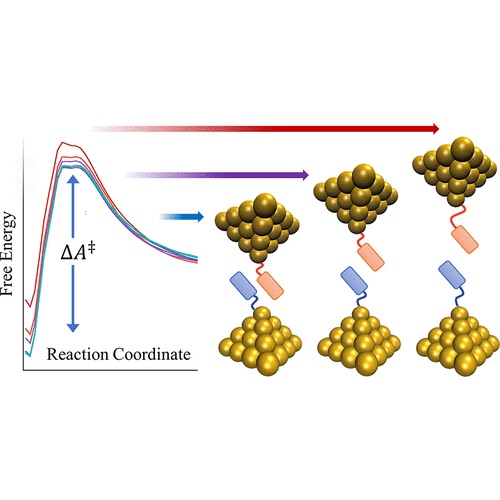
Modeling break-junction experiments: Reproducibility in break-junction experiments relies on measuring the conductance on thousands of freshly formed junctions and performing statistics. The resulting conductance histograms are highly reproducible, and their width is believed to arise because of the uncontrollable variations in junction configurations between experiments. While most simulation efforts focus on a few representative conformations, our group considers all possible conformations and the statistics of junction formation and rupture (see 3 and 4). Contrary to conventional wisdom, these atomistic simulations have revealed that conductance changes during mechanical manipulation is the key physical aspect that needs to be controlled to narrow the width of the conductance histograms (see 4).
Transport beyond Landauer: The central formula in molecular electronics is the Landauer formula which supposes that transport is in steady state and due to quantum tunneling. We are investigating the limits of this formula in thermal environments where the decohering interactions can lead to transport paths that go beyond Landauer (see 5).
Chemical Reactivity: A nascent frontier in molecular electronics is the possibility to investigate chemical reactions. We are studying how how reactivity changes when molecules are nanoconfined in the context of molecular junctions and investigate the fundamental limits of this platform to investigate chemical reactivity at the single-entity limit (see 6 and 7).
Force-Conductance Spectroscopies: Our hypothesis is that force and conductance measurements provide complementary information about the behavior of single-molecules. We have studied this correlation to investigate conformational dynamics (see 1, 2 and 8), hydrogen-bonding (see 9), and propose mechanically-activated molecular switches (see 10 and 11).